Filter BAM/SAM files by insert size
In this blog post, we will talk about what is the insert size in Illumina sequencing reads, and how to find it in SAM/BAM files, and how to filter BAM files with it.
Background
In an Illumina sequencing run, either single-end sequencing (SE) or paired-end sequencing (PE) can be used. In any case, DNA is chopped into small fragments, ligated with adapters at both ends, and sequenced either from one end (SE) or from both ends (PE). For PE reads, insert size refers to the fragment size (excluding the adapters). The name insert, as pointed out by the great blog post Paired-end read confusion - library, fragment or insert size?, comes from the cloning era. It refers to the piece of DNA inserted between the adapters.
Therefore, the insert size includes forward and reverse read as well as the unknown gap between them. The unknown gap can be called the inner mate distance.
It can be visually represented in the following way (discovered from biostars.org):
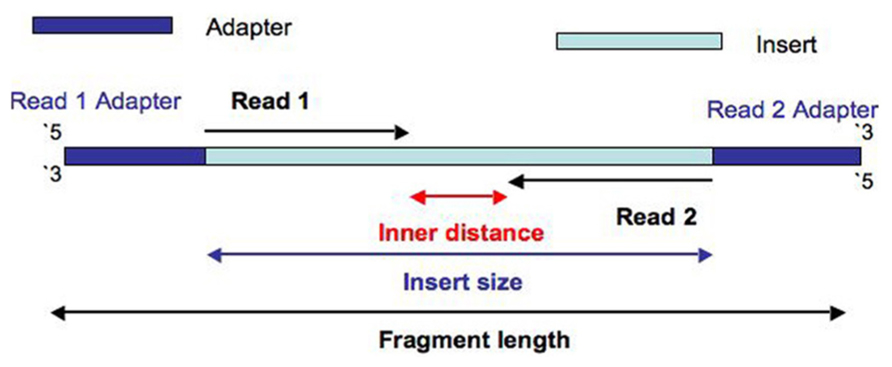
How to find out insert size from a BAM/SAM file or filter the file by it?
The ninth column of a SAM file, observed Template LENgth (TLEN), can be used as an approximate of the fragment length. It is approximate because as documented in the SAM file specification, the exact definition of mapping starts and ends are specific to implementations.
Below, we go through the procedure to collect insert sizes and to filter a BAM file with the insert size. We assume that there is a BAM file named file.bam1.
Example 1: list insert sizes, one line per read
Say we want to plot the distribution of insert sizes in a BAM file, considering only the first pair of the properly mapped pair (flag -f66, see Decoding SAM flags).
samtools view -f66 file.bam | cut -f 9 > insert-sizes.txt
Why there are negative insert sizes?
Using the command above, negative insert sizes can happen. The reason is that the first read is mapped to the reverse strand. In this case, the insert size of the second read is positive.
To report the absolute values, use the following command:
samtools view -f66 file.bam | cut -f9 | awk '{print sqrt($0^2)}' > insert-sizes.txt
The only additional trick is that the awk command takes the absolute value.
Now, you can use your preferred tool, for instance R or Python or even Excel, to visualize the distribution of insert sizes, or to get summary statistics.
Example 2: filter by insert sizes
Say we want to only keep reads of insert sizes between 200 and 500 from a BAM file and write the reads that fulfil the condition to another file.
samtools view -h file.bam | \
awk 'substr($0,1,1)=="@" || ($9>= 200 && $9<=500) || ($9<=-200 && $9>=-500)' | \
samtools view -b > is-200-500.bam
Explanations:
-hinsamtools view: export SAM file headers- In
awk, thesubstrfunction is used to keep header lines, and the rest two condition specify forward and reverse reads with the desired insert sizes, respectively - Last but not least,
samtools view -bis called to write the filtered reads into new a BAM file.
Now, you can load the BAM file into genome browsers, or perform down-stream analysis, for instance feature counting, with it.
Limitation
The insert size estimation using this simple method has an limitation: if the RNA-seq reads were mapped against genomes (especially eukaryotic genomes), instead of transcriptomes, the reported insert size can be huge, up to thousands or even more bases.
It happens because mapping reads to genomes requires that the read aligner splits the reads and map sub-reads to the genome, because in eukaryotic and especially mammalian genomes, two exons can be separated by an intron of hundreds or even thousands nucleotides. This is why estimated insert sizes from genome-mapped SAM/BAM files can be misleading.
There are at least two solutions:
- Re-map the reads to the transcriptome and perform the analysis above. This will avoid the problem of including introns in the insert sizes, because transcriptome sequences do not include introns. The advantage is that it will make the estimates more accurate. The solution comes with a computational and time cost though to remap the data.
- Manually exclude very large inserts, say anything above 1000. The number needs to be determined by the protocol and the need.
An example of solution #2 would be:
samtools view -f66 file.bam | \
cut -f9 | \
awk '{print sqrt($0^2)}' | awk '$0<1000'
Conclusion
Insert size refers to the fragment length consisting of forward and reverse reads and the un-sequenced gap between the paired reads. It is possible to use samtools and command-line tools such as awk and cut to collect insert sizes or to filter BAM/SAM files.
-
To try these commands, it may be useful to sub-sample a big BAM file into a smaller one. See the
-soption ofsamtools viewhow to do that. For instance,-s 35.1will use 35 as a random generator seed and sub-sample 10% of the reads. ↩