Nucleosome positioning and its prediction
Nucleosome positioning is an important factor determining whether a gene is actively transcribed. Together with transcription factor binding, it determines transcriptional behaviour.
- Structure of nucleosome
- Why nucleosome is important
- Experimental approaches to quantify nucleosome positioning
- Software that I found to predict nucleosome binding
- Conclusions
Structure of nucleosome
DNA in our genome, like in genomes of other eukaryotic species, does not exist in a linear form, neither does it form random folding or coiling. Instead, they form nucleosomes together with histone proteins. Each nucleosome consists of 147 DNA base pairs and eight histone proteins (a histone octamer, octo- is a prefix representing in both Latin and Greek the number 8). The way DNA wound on nucleosomes is often described as ‘beads on a string’.

The diameter of a nucleosome is about 11 nanometer (1 nanometer=\(10^{-9}\) m). If DNA in a human cell exists in a linear form, it will be about two meters from one end to the other. Thanks to their compact organization into nucleosomes and higher-order structures, DNA can be fit into human cells, which is about 100 micrometre (\(10^{-6}\) m) in diameter. In fact, chromosomes, which consist of DNA and proteins, takes only a small proportion of it (in diameter of a few micrometer).
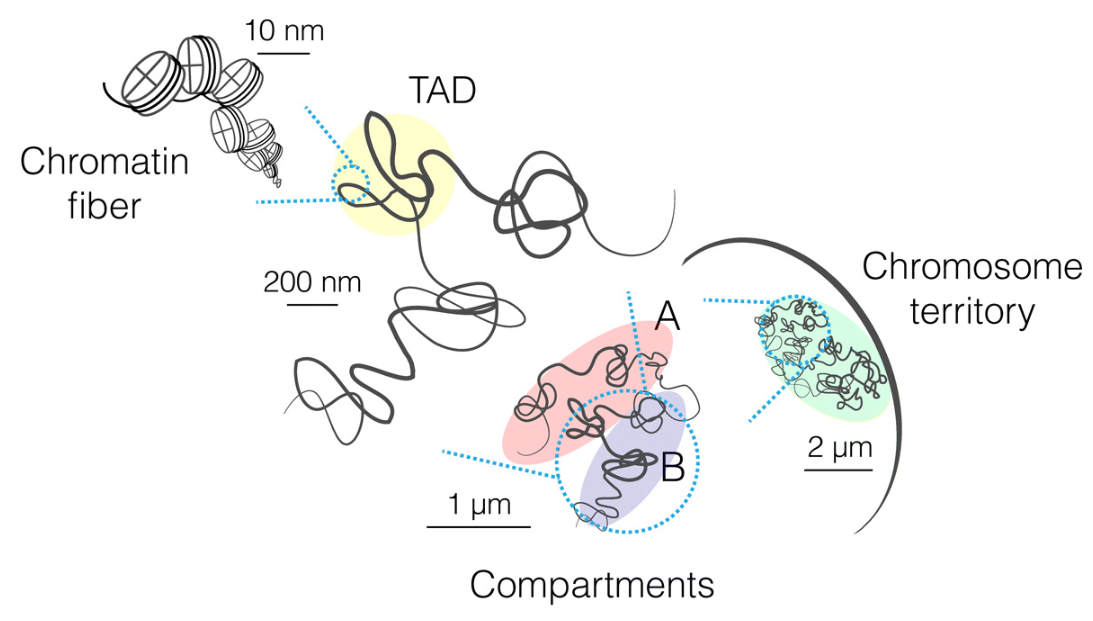
Higher-order structures based on nucleosome
Nucleosome is the first layer of the organization of our chromosomes. They are the foundation of higher layers, which include:
- Chromatin fibre. The beads-on-a-string structure coils into a about 30 nm diameter helical structure known as the 30nm fibre or filament. Histone protein H1 is required for this step. Along the chromatin fibre, the density of nucleosomes varies, depending on and influencing gene regulation.
- Chromatin loops. Chromatin fibre further coils and forms loops, known as chromatin loops.
- Topologically associated domains (TADs). Chromatin loops fold at a sub-megabase scale (1 megabase, or 1Mb, is 1,000,000 bases), and they form higher domains of preferential interactions, known as topological associated domains (TADs), with the size of a few hundred nanometer.
- Compartment. On the chromosomal scale, chromatin in TADs is segregated (which means setting apart from each other) into two major compartments: the active “A” compartment and repressed, inactive “B” compartment.
- Territories. Finally, entire chromosomes segregate into distinct regions in the nucleus occupy distinct sub-nuclear chromosome territories.
The following figure from Principles of genome folding into topologically associating domains by Szabo et al. (Science Advances, 2019) offers a graphical overview of the hierarchical organization.
Another good resource to get an first impression of DNA packaging and how nucleosome and chromatin contribute to the process is available Scitable, an education resource of the company Nature Publishing Group.
Why nucleosome is important
We learned above that nucleosomes are important to package DNA strands densely. And we learned that they regulate gene expression. In fact, a model of gene transcription regulation proposed by Eran Segal and Jonathan Widom (Nature Review Genetics, 2009) postulates that transcriptional regulation can be explained by an ‘equilibrium competition’ between nucleosomes and other DNA-binding proteins such as transcription factors.
It was a bit surprising to me when I read it for the first time. Human histones contain five subgroups (H1, H2A, H2B, H3, and H4), and contain only 110 genes (see this page maintained at genenames.org for an overview of them). Almost half of them are encoded in a large cluster on human chromosome 6. In comparison, there are more than 1,600 known or likely human transcription factors (The Human Transcription Factors, Lambert et al., Cell, 2018). In addition, interactions and collaborations between histones seem much less complex than the collaborations between transcription factors. It was surprising to me that so few histones determine gene expression together with so many transcription factors.
On the other hand, though, the importance of nucleosomes is not surprising. As we saw above, nucleosomes are the foundation of the chromosomal organization. A transcription factor, in order to drive expression of a gene, must compete with the nucleosome to access the DNA, because a piece of DNA can hardly be in a nucleosome and bound by a transcription factor at the same time (exceptions may exist, though here is the general rule), which we call the steric hindrance, the phenomenon that spatial arrangement of atoms and particles affecting chemical reactions.
Nucleosome and transcription factors
It is widely believed now that nucleosomes and transcription factors compete with each other to drive expression. Segal and Widom proposed to consider all possible configurations of nucleosome and transcription factor binding, and to use the weighted sum to predict transcription activity. This model, for instance, can be used to explain noise in gene expression levels.
We use the term transcriptional noise to indicate the variability in the
transcriptional rate of genes across different cells from a homogeneous cell
population. In case these cells are in a steady state, the variability is also
reflected on the mRNA expression levels. TATA sequences, a DNA sequence with
a core sequence of 5'-TATA-3' found in the promoter regions of many
genes which helps recruiting RNA polymerase to a promoter, predict high levels
of noise, maybe by reinitializing transcription. In contrast,
nucleosome-disfavouring sequences predict low levels of noise. This may be
explained by that lack of competition by nucleosome and therefore a better
accessibility of transcription factors.
Interestingly, these genes with nucleosome-depleted regulatory regions and low transcriptional noise can switch between inactive and active states of gene expression more rapidly, partially because their expression needs less and is less targeted by chromatin remodelers. Chromatin remodelers are protein complexes that alter the structure of chromatin, which consume much energy and are, therefore, ‘expensive’.
Factors contributing to nucleosome positioning
DNA binding to nucleosomes is a dynamic process that shows sequence preference. In another word, some DNA sequence is more likely to be in a nucleosome than other sequences. This is similar with transcription factors, which also show sequence-preference binding. But there are important differences between the histone-DNA and transcription factor-DNA binding.
Because many transcription factors have a much shorter binding sequence (a few nucleotides long), which can appear many times in the genome, the binding patterns of transcription factors to genome in living cells (in vivo) can show much more randomness, among other because the number of transcription-factor proteins may be fewer than the available sites.
This is not the case for histone binding of DNA. On one side, the preferred binding sequence motifs are much longer for histones. On the other side, there are usually abundant histones available to bind to their preferred sequences.
Besides DNA sequence, nucleosome binding is influenced by the following factors:
- ATP-dependent nucleosome remodelling enzymes,
- Non-histone DNA-binding proteins such as transcription factors,
- The transcription machinery, including the pre-initiation complex and elongating RNA polymerase II.
These factors are reviewed by Struhl and Segal and by Chereji and Clark.
Experimental approaches to quantify nucleosome positioning
Four commonly experimental approaches to quantify nucleosome positioning are DNase-seq, FAIRE-seq, ATAC-Seq, and MNase-Seq. Here I focus on ATAC-Seq and MNase-seq to illustrate the principles. A brief comparison of the four approaches is available on Wikipedia.
ATAC-seq uses a hyperactive transposase to tag linkers, which are nucleosome-free DNA segments, for sequencing. The transposase is loaded with sequencing adaptors, and it cuts linker sequences and adapts sequencing adaptors at the same time. Thus it can be used to profile sequences that are not occupied by nucleosomes. The protocol was described in Buenrostro et al. (Nature Methods, 2013).
MNase-seq uses Micrococcal nuclease to degrade linker DNA, thereby preserving the DNA sequences wrapped in nucleosomes, which are then sequenced. Several groups contributed to its development.
Software to analyse such data include ATACseqQC in Bioconductor and Nucleosome Dynamics reported by Buitrago et al. (NAR, 2019).
Software that I found to predict nucleosome binding
Complementary to experimental approaches, in silico tools predict nucleosome positioning by using DNA sequences alone.
I found two open-source software packages for this purpose.
NuPop
Xi et al. published in 2010 the software NuPop (Xi et al., BMC Genomics 2010). It uses a duration Hidden Markov Model for nucleosome position prediction. An interesting feature is that it explicitly models the linker DNA length. The core model was trained using yeast data, and the model parameters are scaled to reflect genome structures of other species, including the human genome.
The software was written in Fortran and a R package as a wrapper is available. It is available in Bioconductor](https://www.bioconductor.org/packages/release/bioc/html/NuPoP.html), as well as on GitHub jipingw/NuPoP.
For those who want to see the parameters of the models, it seems that the binary
file sysdata.rda contains
them.
To those who are interested in differential splicing, the paper of Xiong et
al., The human splicing code reveals new insights into the genetic
determinants of disease, published in 2015 in Science may be familiar. In
fact, the authors used NuPop to predict nucleosome positioning in their study.
LeNup
A recent piece of software to learn sequence-based features of nucleosome positioning and to predict nucleosome positioning patterns is LeNup (Zhang et al., Bioinformatics, 2018).
It uses a convolutional neural network to predict nucleosome positioning in Homo sapiens and other species. The open-source software is written in Python and Lua, and is available on GitHub at biomedBit/LeNup.
Conclusions
I am fascinated by how nucleosome and transcription factor compete to regulate gene expression. The summary above is my notes of only a tiny proportion of what we know about nucleosomes and how we may study them.
If we aim to link gene expression with either disease aetiology or drug-treatment response with better in silico models, we may benefit from integrating our knowledge of and uncertainty about nucleosome and transcription factors into mathematical and computational models describing gene expression behaviours.